Electronic Excitations: Molecular Spectra and Structures of Excited States
 Electronically excited states have usually a complicated
electronic structure, and usually also the molecular geometry is different from that in the ground state and difficult
to predict. Chemical calculations are therefore an important tool for
the understanding of molecular spectra and the photochemical
reactivity of molecules. Modern electronic structure methods, as e.g.
density functional and coupled-cluster response methods allow today
to investigate with ab initio methods excited
states of relatively large molecules, to predict accurately their spectra, and to determine the equilibrium structures
of excited states. In our group we apply for these investigations (in
addition to TDDFT) mainly the RI-CC2 approach, which has been
developed in the group and is well-suited for applications on medium
sized and large organic chromophores (up to ca. 100 atoms). Examples
which have been studied with this approach in our group are:
Electronically excited states have usually a complicated
electronic structure, and usually also the molecular geometry is different from that in the ground state and difficult
to predict. Chemical calculations are therefore an important tool for
the understanding of molecular spectra and the photochemical
reactivity of molecules. Modern electronic structure methods, as e.g.
density functional and coupled-cluster response methods allow today
to investigate with ab initio methods excited
states of relatively large molecules, to predict accurately their spectra, and to determine the equilibrium structures
of excited states. In our group we apply for these investigations (in
addition to TDDFT) mainly the RI-CC2 approach, which has been
developed in the group and is well-suited for applications on medium
sized and large organic chromophores (up to ca. 100 atoms). Examples
which have been studied with this approach in our group are:
the transition energies to the lowest excited states in trans- and cis-azobenzene and their change on substitution in different positions with a variety of functional groups
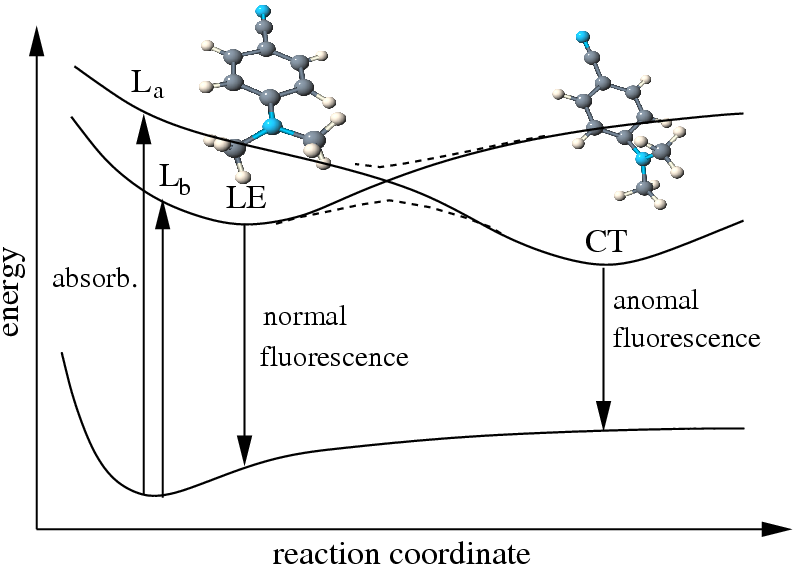
the equilibrium structures of the lowest excited states of 4-(dimethyl-amino)-benzonitrile (DMABN) and the related NMC6 and NTC6, which are prototype molecules for the investigation and understanding of the so-called dual fluorescence phenomenon
-
UV/VIS spectra of chlorophyll chromophores and their change with chemical modification and/or complexation and the influence of the chlorophyll—chlorophyll interaction in e.g. natural photosystems.
Additionally,
-
in cooperation with the group of Prof. Dr. Samuel Leutwyler in Bern we benchmarked the accuracy of quantum chemical methods for the prediction of 0–0 transitions of aromatic organic molecules
-
in cooperation with the group of Prof. Dr. Patrick Nürnberger we studied the ultrafast dynamics of the aromatic triazene berenil after photexcitation.